Rail-to-rail OpAmps
It is no mystery that the good old LM324 is a bit outdated already. It was a beast in it's day: cheap, versatile and low-power. However, its relatively simple architecture is somewhat flawed near the supply rails (and especially if you are using single supplies). Each opamp in the LM324 presents an output stage composed of a Darlington pair (see Figure 1).
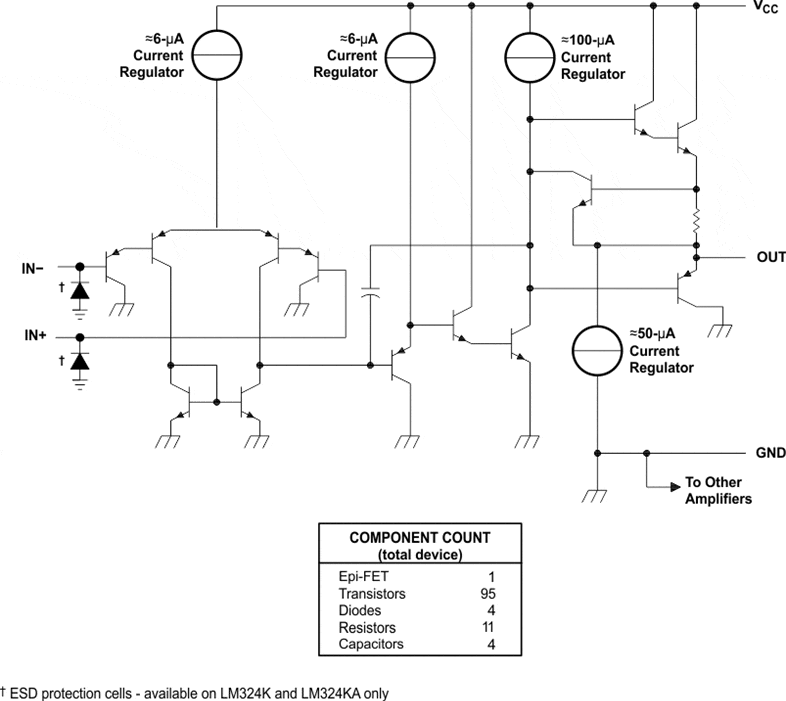
Figure 1 - Schematic diagram of the LM32x
This Darlington pair configuration (see Figure 2) is essentially a single transistor (one Base, one Collector and one Emitter). The reason to stick it into the output stage is mainly to provided for a higher current gain (since βDAR ≈ β1·β2) and to decrease the output impedance of the stage.

Figure 2 - Simple Darlington pair
However, while doing so one now has to overcome the Collector-Emitter voltage of the first transistor, and Base-Emitter voltage of the second in the pair. Run through the math and turns out you are 0.2 - 0.1 V under the high and over the low rails (since VC2 > VB2). This is commonly known as output saturation. (The input voltage range is tied as well by the input stage, but that is a topic for another post).
You can, of course, drive the supply rails a over the minima of your system by widening the supply rails from -VCC to +VCC and ensuring that |VOUT|< |VCC - 0.2|. But, what happens when you do not have a negative supply rail and need that 0.1 V output?
You have two options here:
- You can use an additional chip to create a small negative bias rail. Chips like the LM7705 are designed to do this specifically. However, they are usually sold in batches of 2500 units, and are a bit out of the hobbyist hand.
- Use a rail-to-rail opamp. Several newer designs (both BJT and CMOS) have come up with stages that allow them to come closer of the supply rails (typically within tens of millivolts, although this can vary with the bias). Some cheap chips that can achieve this are the MC34074 (100 mV at VCC from 0 to 5 V), MC33174 (50 mV at VCC from 0 to 5 V), MC33204 (150 mV at VCC from 0 to 5 V), TLV2374 and TLC274 (50 mV at VCC from 0 to 5 V), TLV2464 (200 mV at VCC from 0 to 5 V), LM6134 (75 mV at 24 V) or LMC6484.
In order to find at how much does the opamp saturate when driven to the rail, look in the datasheet for "Output Voltage Low" section and take into account the test characteristics (since different bias and loads will surely affect the output of the opamp).
In my case, the solution was to add a dual power supply, but sometimes a rail-to-rail opamp is very useful.
Alexander M. Pasek
Biomedical Engineer